Jan 25, 2021
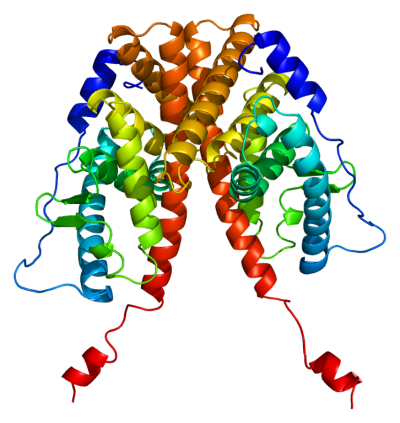
Proteins play an essential role in determining structural components of body tissue, enzymes, and antibodies. Understanding how cells determine which proteins to produce is the key to preventing disease, cellular mutations, and more.
A gene must be first expressed before its protein product is produced. Now, with advancements in computational science, researchers are asking if we can computationally identify the mechanism which decides which genes express themselves in a cell.
The answer to this billion-dollar question lies in predicting genetic regulatory networks using large-scale single cell gene-expression data.
School of Computational Science and Engineering (CSE) Assistant Professor Xiuwei Zhang is the recipient of a $400,000 National Science Foundation grant supporting the creation of new computing methods that aims to do just that.
“We know that if we detect expression for a gene then it is likely that its proteins are also present. Since experimentally measuring protein abundance in cells is very difficult, researchers look to gene regulatory networks to understand which proteins are present instead,” she said.
A gene regulatory network is a directing graph which shows, out of tens of thousands of genes, which genes are controlling other genes.
“A common theory people use about molecular biology is that one gene corresponds to one mRNA and then corresponds to one protein. And most of the existing work to learn the gene regulatory networks also use this theory. However, this theory is over-simplified, and the fact is that one gene can correspond to multiple mRNAs, thus multiple proteins,” said Zhang.
This is where Zhang’s research breaks from traditional approaches and considers this one-to-many relationship in its gene regulatory networks.
“Now since one gene corresponds to multiple isoforms, in our gene regulatory networks, the nodes are isoforms instead of genes, which can provide a more accurate representation of the actual regulatory mechanism in cells,” she said.
According to Zhang, recent advances in single cell RNA-sequencing technology have introduced new opportunities to infer high-quality regulatory networks at this level, but also pose new computational challenges
In response to these challenges, a method for developing a transcript assembler that can quantify the expression level an isoform is needed to build an accurate and scalable regulatory network. This part of the work is led by Zhang’s collaborator, Pennsylvania State University Assistant Professor Mingfu Shao.
Another challenge for the researchers to access network accuracy has to do with cell ordering which plays a major role in inferring an accurate network. Depending on the level of error, cell ordering will determine whether a regulatory network’s predictions are accurate. To ensure this new network’s predictions are accurate, Zhang has the robust goal to create a method that can perform cell ordering and network inference simultaneously.
Ultimately, the new methods will be used in the field of immunology to study cellular mechanisms in steroid-producing cells with collaborators at Cambridge University.
“This is very important for many biological events such as if disease happens during embryo development or to the immune system. It is our goal to be able to see from data that the level of an expression of a certain gene is not normal and then trace the problem through the regulator network. Once this is done, we can begin targeting the upstream genes for drug or vaccine development,” said Zhang.
Related Media
-
Network Inference