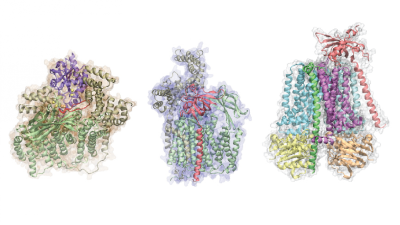
A computational tool developed to predict the structure of protein complexes – the molecular machinery that makes life possible – is lending new insights into the biomolecular mechanisms of their function.
Apr 18, 2022
From the muscle fibers that move us to the enzymes that replicate our DNA, proteins are the molecular machinery that makes life possible.
Protein function heavily depends on their three-dimensional structure, and researchers around the world have long endeavored to answer a seemingly simple inquiry to bridge function and form: if you know the building blocks of these molecular machines, can you predict how they are assembled into their functional shape?
This question is not so easy to answer. With complex structures dependent on intricate physical interactions, researchers have turned to artificial neural network models – mathematical frameworks that convert complex patterns into numerical representations – to predict and “see” the shape of proteins in 3D.
In a new paper published in Nature Communications, researchers at Georgia Tech and Oak Ridge National Laboratory build upon one such model, AlphaFold 2, to not only predict the biologically active conformation of individual proteins, but also of functional protein pairings known as complexes.
The work could help researchers bypass lengthy experiments to study the structure and interactions of protein complexes on a large scale, said Jeffrey Skolnick, Regents’ Professor and Mary and Maisie Gibson Chair in the School of Biological Sciences and one of the corresponding authors of the study, adding that computational models such as these could mean big things for the field.
If these new computational models are successful, Skolnick said, “it could fundamentally change the way biological molecular systems are studied.”
Primed for Protein Prediction
Created by London-based artificial intelligence lab DeepMind, AlphaFold 2 is a deep learning neural network model designed to predict the three-dimensional structure of a single protein given its amino acid sequence. Skolnick and fellow corresponding author, Mu Gao, senior research scientist in the School of Biological Sciences, shared that the Alphafold 2 program was highly successful in blind tests occurring at the 14th iteration of the Community Wide Experiment on the Critical Assessment of Techniques for Protein Structure Prediction, or CASP14, a bi-annual competition where researchers around the globe gather to put their computational models to the test.
“To us, what is striking about AlphaFold 2 is that it not only makes excellent predictions on individual protein domains (the basic structural or functional modules of a protein sequence), but it also performs very well on protein sequences composed of multiple domains,” Skolnick shared. And so with the ability to predict the structure of these complicated, multi-domain proteins, the research team set out to determine if the program could go a little further.
“The physical interactions between different [protein] domains of the same sequence are essentially the same as the interactions gluing different proteins together,” Gao explained. “It quickly became clear that relatively simple modifications to AlphaFold 2 could allow it predict the structural models of a protein complex.” To explore different strategies, Davi Nakajima An, a fourth-year undergraduate in the School of Computer Science, was recruited to join the team’s effort.
Instead of plugging in the features of just one protein sequence into AlphaFold 2 per its original design, the researchers joined the input features of multiple protein sequences together. Combined with new metrics to evaluate the strength of interactions among probed proteins, their new program AF2Complex was created.
Charting New Territory
To put AF2Complex to the test, the researchers partnered with the high-performance computing center, Partnership for an Advanced Computing Environment (PACE), at Georgia Tech, and charged the model with predicting the structures of protein complexes it had never seen before. The modified program was able to correctly predict the structure of over twice as many protein complexes as a more traditional method called docking. While AF2Complex only needs protein sequences as input, docking relies on knowing individual protein structures beforehand to predict their combined structure based on complementary shapes.
“Encouraged by these promising results, we extended this idea to an even bigger problem, which is to predict interactions among multiple arbitrarily chosen proteins, e.g., in a simple case, two arbitrary proteins,” shared Skolnick.
In addition to predicting the structure of protein complexes, AF2Complex was charged with identifying which of over 500 pairs of proteins were able to form a complex at all. Using newly designed metrics, AF2Complex outperformed conventional docking methods and AlphaFold 2 in identifying which of the arbitrary pairs were known to experimentally interact.
To test AF2Complex on the proteome scale, which encompasses an organism’s entire library of the proteins that can be expressed, the researchers turned to the Summit Oak Ridge Leadership Computing Facility, the world’s second largest supercomputing center. “Thanks to this resource, we were able to apply AF2Complex to about 7,000 pairs of proteins from the bacteria E. coli,” Gao shared.
In that test, the team’s new model not only identified many pairs of proteins known to form complexes, but it was able to provide insights into interactions “suspected but never observed experimentally,” Gao said.
Digging deeper into these interactions revealed a potential molecular mechanism for protein complexes that are particularly important for energy transport. These protein complexes are known to carry hemes, essential metabolites giving blood dark red color. Using AF2Complex’s predicted structural models, Jerry M. Parks, a senior research and development staff scientist at Oak Ridge National Laboratory and a collaborator in the study, was able to place hemes at their suspected reaction sites within the structure. “These computational models now provide insights into the molecular mechanisms for how this biomolecular system works,” Gao said.
“Deep learning is changing the way one studies a biological system,” Skolnick added. “We envision methods like AF2Complex will become powerful tools for any biologist who would like to understand molecular mechanisms of a biosystem involving protein interactions.”
AF2Complex is an open-source tool available to the public and can be downloaded here.
This work was supported in part by the DOE Office of Science, Office of Biological and Environmental Research (DOE DE-SC0021303) and the Division of General Medical Sciences of the National Institute Health (NIH R35GM118039). DOI: https://doi.org/10.1038/s41467-022-29394-2
Related Media
-
Researchers Jeffrey Skolnick and Mu Gao at the Engineered Biosystems Building at Georgia Tech. (Photo: Jess Hunt-Ralston)
-
A 3D rendering of a protein complex structures predicted from protein sequences by AF2Complex. Credit: Mu Gao.
-
The initial development of AF2Complex was done at the Partnership for an Advanced Computing Environment (PACE) computing center of Georgia Tech, pictured here in the Coda Data Center. Credit: Paul Manno/PACE.
-
The Summit supercomputing center at Oak Ridge National Laboratory. Credit: Oak Ridge National Laboratory.